

欢迎光临吉康旅!
发布日期??Ŀ化学药物评价>>综合评价题目讨论与案例分析小分子靶向抗癌药物临床研究策略公司的研究进展,一些抗肿瘤药物新靶点的发现,肿瘤领域的药物开发经历了巨大的发展变化,从传统的细胞毒药物到非细胞毒分子靶向药物的开发,新产品陆续推出上市。2011年FDA批准的7个新药中,有6个是分子靶向药物[1]。由于作用机制不同,小分子靶向药物表现出与传统细胞毒药物不同的安全性和有效性特征,临床研究设计和开发模式也不同。本文回顾分析了近年来获批上市的一些典型案例,如吉非替尼、克唑替尼、埃克替尼等小分子靶向药物的临床开发情况,并探讨了不同类型抗肿瘤药物的临床开发策略。,希望为相关研发人员提供参考。1.传统抗癌药物临床研究模式 抗癌药物遵循一般药物临床研究规则。上市前研究通常分为I期、II期和III期临床试验。I期临床试验的主要目的是评价药物的耐受性和药代动力学,为II期研究推荐给药方案;II期临床试验的主要目的是在小样本中初步观察药物的有效性,同时也观察安全性;III期临床试验在II期的基础上选择目标适应症人群,进一步扩大样本量,以确认前期研究中观察到的获益和风险,为获得上市许可提供充分的证据。
由于肿瘤药物和疾病的特点,抗肿瘤药物的临床研究具有不同于一般药物的特点。例如,由于抗肿瘤药物往往毒性较大,为了避免对健康受试者造成不必要的伤害,I期研究的首次人体试验往往是在癌症患者中进行,而其他领域的药物则多在健康人群中进行。志愿者。. 同时,出于伦理考虑,通常新的抗肿瘤药物应首先在没有有效治疗方法或在现有标准治疗后无效或复发的难治性患者中进行,并在对难治性病例取得积极效果后,然后逐步推进到新治疗患者的一线治疗和辅助治疗。此外,有必要研究该药物的抗肿瘤谱。抗肿瘤药物通常不是对所有肿瘤类型都有效,也不是只对一种肿瘤有效。因此,有必要在早期筛选出敏感的肿瘤类型,以供后期研究使用。一般来说,抗肿瘤药物的疗效和安全性与剂量/方案密切相关,不同的给药方案(如给药间隔和剂量等)可能产生不同的剂量限制性毒性(Dose、DLT)和最大耐受剂量(Dose,MTD)。对于细胞毒类药物,在毒性可以耐受的前提下,提倡尽可能增加剂量,以达到最佳效果。所以,
此外,由于肿瘤单药治疗容易产生耐药性,临床上推荐使用联合治疗。联合应用毒性不完全重叠的化合物或联合应用具有不完全重叠耐药机制的化合物,可以达到可接受的毒性。提高抗肿瘤活性水平的目的。因此,探索联合用药的安全性和有效性也成为早期研究的重要内容。可见,抗肿瘤药物在试验人群和给药方案中的探索过程较为复杂,在长期实践中形成了较为成熟的研究模式,主要来源于传统细胞毒药物的经验。药物。在大多数情况下,遵循此模型:阶段I分为两个阶段,Ia和Ib。首先,对包括多种肿瘤类型在内的难治性肿瘤患者进行单剂量剂量递增耐受试验,以毒性为主要终点,确定单药的MTD。随后进行了联合给药的剂量递增耐受试验,并以毒性作为主要终点确定联合给药的MTD,并推荐了II期剂量。第二阶段也分为两个阶段,IIa 和 IIb。首先,初步研究单一药物对某些潜在有效肿瘤类型的疗效,以客观肿瘤缓解为主要终点,然后研究不同肿瘤类型的不同联合给药方案。功效。在这段时期,Ib 和 IIa 研究的启动时间可能会重叠,每个阶段可能会有多个研究。在前期研究结果的基础上,我们将首先选择最有潜力的敏感肿瘤物种和最佳给药方案进入III期研究阶段,然后对其他敏感肿瘤物种进行一一的III期研究,以扩大研究范围。适用人群,优化给药方案。研究。
2.小分子靶向抗肿瘤药物的临床研究模式不同于传统的细胞毒药物。小分子靶向药物多以膜受体、细胞信号转导通路的组成成分、细胞周期调控蛋白、重要蛋白或因子等参与血管生成的靶点为靶点,具有相对特异性,对正常细胞的毒性相对较小。由于其抗肿瘤作用可能是抑制肿瘤细胞生长或防止转移,而不是杀死肿瘤细胞,因此客观缓解率可能不是衡量抗肿瘤活性的合适指标。在大多数情况下,此类化合物的暴露(给药)时间需要延长,并且经常需要连续给药而不是间歇给药,以实现对靶标的持续抑制。以上特点使得小分子靶向药物的临床应用研究模式有别于传统的细胞毒药物,尤其是在临床研究的早期阶段。在研究人群的选择、剂量/方案的确定和终点指标的选择上存在差异。例如,由于毒性较低,有可能选择健康人进行一些早期试验;在探索剂量和方案时,毒性可能不是一个合适的终点。需要根据药代动力学和药效学(抑制特定靶点而不是肿瘤缩小)的结果综合确定;为了增加后续研究力度,可能需要对疗效进行早期评估,甚至是随机对照设计,因此有时很难严格区分 I、II 和 III 期。因此,此类药物的早期临床研究变得更加灵活、多样、丰富和复杂,以追求更高效、更准确的目标人群定位和更准确的剂量,提高后期研究的信心。
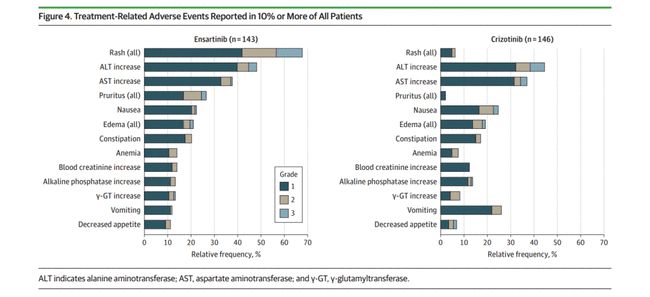
回顾近年来开发的小分子靶向药物的临床研究设计,早期研究反映的一个共同趋势是:有效剂量组增加病例数,并在推荐的II期剂量下继续加入扩展研究,并直接进入II期阶段,该阶段将I/II期研究的目标与两阶段模型相结合。在研究中获取尽可能多的信息,以提高早期研究的效率,增强药物进入后期的信心。以辉瑞公司于2011年获得FDA批准的克唑替尼()为例。它的第一个 I 期研究是一项两阶段、不受控制的剂量递增研究(n = 119 例)[2]。研究内容包括单项和多项耐受性研究(单项和多项研究之间设置7天的导入期)、单项和多项药代动力学研究、药物相互作用研究、食物效应研究、每日两种方案的比较研究一天一次和两次。疗效评价指标包括生物标志物、肿瘤缓解(如ORR、DR)和生存时间相关指标(如PFS、OS),在本研究中同时实现了传统的I/II期研究目标(见图1).注:MTD/RP2D:最大耐受剂量/II期推荐剂量图1.克唑替尼-研究设计小分子靶点 药物研发中一个非常重要的方面是证明其具有理想的“靶点”效果. 单次和多次药代动力学研究、药物相互作用研究、食物效应研究、每日 每日一次和两次两种方案的比较研究。疗效评价指标包括生物标志物、肿瘤缓解(如ORR、DR)和生存时间相关指标(如PFS、OS),在本研究中同时实现了传统的I/II期研究目标(见图1).注:MTD/RP2D:最大耐受剂量/II期推荐剂量图1.克唑替尼-研究设计小分子靶点 药物研发中一个非常重要的方面是证明其具有理想的“靶点”效果. 单次和多次药代动力学研究、药物相互作用研究、食物效应研究、每日 每日一次和两次两种方案的比较研究。疗效评价指标包括生物标志物、肿瘤缓解(如ORR、DR)和生存时间相关指标(如PFS、OS),在本研究中同时实现了传统的I/II期研究目标(见图1).注:MTD/RP2D:最大耐受剂量/II期推荐剂量图1.克唑替尼-研究设计小分子靶点 药物研发中一个非常重要的方面是证明其具有理想的“靶点”效果.
整个研发过程其实就是一个寻找和验证目标的过程。一系列问题需要回答:药物对靶点有影响吗?对目标的影响是否会产生后续的生物学效应?哪种给药方案对目标的影响最大?影响疾病预后的关键因素有哪些?它对具有某些分子特征的人更有效吗?这就需要在早期研究中尽可能多地收集与靶点相关的信息,包括保留生物样本进行生物标志物分析,对早期研究中获得的信息进行回顾性或前瞻性探索和分析以获得相关提示,然后设计相应的研究进行进一步验证。近年来部分病例的开发经验表明,早期识别可预测疗效和预后的生物标志物,选择具有一定特征的优势人群进行临床研究可能是导致试验成功甚至加速的途径之一。临床开发过程。以阿斯利康生产的吉非替尼为例。该产品于 2003 年通过了 FDA 的第二阶段试验(test)。2005 年,由于承诺的上市后 III 期测试(ISEL 测试),它被报告为阴性结果。FDA限制适应症,遭受重创。好在阿斯利康并没有因此放弃,因为亚组分析显示亚洲人、女性、腺癌和非吸烟者的结果更好。在2008,在亚洲非吸烟者中进行了一项试验。最终获得了积极的结果,并获得了欧盟的批准上市。
经过10多年的研究和分析,已经确定EGFR突变是决定服用EGFR-TKI患者疗效和预后的重要因素。亚洲人、女性、腺癌和非吸烟者是EGFR突变率高的人群,所以试验成功了,而ISEL试验因为没有选择受益于优势人群而被无效人群“稀释”。2011 年版的 NCCN 指南推荐它作为 EGFR 突变的首选药物。相比之下,辉瑞的克唑替尼要幸运得多。2006 年,开始了一项临床研究。2007年,一项研究发现EML4?ALK融合基因是非小细胞肺癌肿瘤形成的另一个独立关键驱动因素[3],随后修订了I期研究的研究人群进入标准。第二阶段特意选择ALK基因易位或倒位的非小细胞肺癌患者。正如预期的那样,观察到了出色的疗效 [2 (] 见图) 1),然后在该人群中进行了 II 期研究 (n=136))。这两项研究成为支持该产品在美国上市的关键研究证据。从第一次临床试验到提交上市申请仅用了5年时间,大大缩短了开发周期。由于已经确定与临床疗效相关的生物标志物毕竟是有限的,并不是所有的药物都能有克唑替尼的运气,所以首次筛查在人群中进行临床试验并不普遍,
3.中国对小分子靶向药物临床研究策略的探索与全球研发趋势一致。目前国内抗肿瘤新药的研发也集中在分子靶向抗肿瘤药物上,其中小分子酪氨酸激酶抑制剂(TKI)是最热门的研发。2005年至2012年,此类药物申报的新化合物总数达到28个,占所有抗肿瘤新药的1/2以上。这些药物大多是“me-too”药物,在现有上市药物或在研药物的基础上进行结构改造,具有相似的安全性和有效性特征。不同情况下产品的临床研发策略应该是不同的。国内目前在研的新化合物存在三种情况:1)新靶点,无药物开发经验或使用同一靶点;2) 已知靶点,具有相同靶点药物开发经验,但尚未发现与疗效和预后相关的生物标志物;3) 已知靶点,具有相同靶点药物开发经验,并已发现可预测疗效和预后的生物标志物。对于有新靶点的药物,一切还是未知,需要遵循一般的研究规律,可能还需要更多的研究,但对于已知靶点的药物,要充分借鉴已有经验,认真研究分析,根据研究资料国内外相同靶点药物,根据产品特点和实际情况,选择适合自身产品的临床研究模式,少走弯路。以2011年获得SFDA批准的浙江北大生产的盐酸埃克替尼为例:该产品是罗氏公司在2004年获得FDA批准的厄洛替尼(?)基础上,经结构改造获得的。, 是一种表皮生长因子受体 (EGFR) 酪氨酸激酶抑制剂。
针对同一目标的上市产品包括上述阿斯利康(易瑞沙?)生产的吉非替尼。可以说这款产品的开发有更多的经验可以借鉴。盐酸埃克替尼的临床研究于2005年底开始,2010年底结束,历时5年完成5个I期、2个I/II期、1个III期研究(见表1)从表1可以看出,申办方基本沿用了目前此类药物的临床研究思路,同时也考虑了me-too产品本身的特点,充分借鉴了吉非替尼和厄洛替尼的现有信息进行比较的思路。后两者贯穿始终。例如,进行了足够的早期探索性研究;选择健康志愿者进行单剂量耐受和药代动力学相关研究,对癌症患者进行多剂量耐受。及药代动力学研究;根据产品的药代动力学特征,参考同一靶标药物的临床给药方案,探讨给药间隔;基于 I 期剂量递增试验的潜力,采用了 I/II 期联合设计。扩大病例观察到有效安全剂量水平,达到二期研究目标;参照同一靶标药物的临床适应症,未开展不同肿瘤类型的探索性研究,直接选择靶标适应症人群;关键研究是基于相同的靶点药物作为对照,采用二、三期联合设计,制定提前终止标准。同时,按照现行国际惯例加强试验质量控制,包括采用双盲双模拟,应用IWRS进行随机化,设立独立的疗效评价委员会,数据监测委员会、专家指导委员会、数据管理系统介绍、临床试验注册等。
虽然还存在一些统计设计、数据收集和实施方面的问题,对结果的解读有一定的影响,但仍是现阶段我国自主研发药物开发的典型成功案例。这些问题是申请人在上市后要求的。进一步的研究。表 1:盐酸埃克替尼完成的试验总结 研究类型研究设计病例数研究站点 1 在健康中国男性志愿者中评估单次口服盐酸埃克替尼片 A 的安全性和耐受性,I 期,单中心,随机,双盲、安慰剂对照研究(2006.12-2007.3) 在中国健康男性志愿者中)A 期,单中心,

比如上面提到的盐酸埃克替尼,虽然已经有两个上市化合物作为参考,但已经不可能重复任何一种药物的研究设计。在制定临床研究计划和方案时,我们经常要对一些关键问题做出决策。例如,第一次人体试验的受试者群体、健康志愿者或肿瘤患者的选择?选择具有某些分子特征的人还是非选择性的人?选择市场上具有相同目标药物适应症的人群还是开发新的适应症?还有参考药物的选择。选择现有的标准化疗作为对照?还是选择已经上市的相同靶点药物作为对照?此外,在各个阶段的给药方案探索和终点指标的选择上都会遇到类似的问题。具体问题需要具体分析,这里很难总结出适用于所有药物的指南。重要的是应首先确定总体发展战略,这往往对程序设计中的具体问题产生关键影响。通常有两种策略:一是完全遵循市场上或在研现有产品的研究思路,选择与开发目标相同的适应症人群,证明其产品不比现有产品差或好。这种策略由于借鉴了早期的经验,比较简单快捷,开发风险小。缺点是竞争激烈,市场份额不断缩小。如果您从研究中的产品中学习,您还需要考虑正在使用的产品的失败风险以供参考。二是选择已经上市或产品尚未开发的适应症人群,根据自身产品特点寻找新的突破口。显然,这种策略风险相对较大,但可能开辟新市场,也有利有弊。并根据自身产品特点寻找新的突破口。显然,这种策略风险相对较大,但可能开辟新市场,也有利有弊。并根据自身产品特点寻找新的突破口。显然,这种策略风险相对较大,但可能开辟新市场,也有利有弊。
还有第三种选择,就是先从最容易开发的适应症入手,然后在市场之后探索新的适用人群来拓展市场。申办方仍应根据自身产品特点和现有条件选择最合适的研究策略。还是以盐酸埃克替尼为例。在临床前研究中,本品显示出与吉非替尼和厄洛替尼相似的抗肿瘤活性,毒性稍低,但半衰期短,代谢广泛是其缺点。因此,最好尽快确定本品的人体药代动力学特性。关键是申办者首先选择健康志愿者进行单剂量耐受和药代动力学研究,并以安慰剂组为对照,以获得明确的安全性和药代动力学数据;然后直接选择吉非替尼,厄洛替尼已获批用于适应症人群——非小细胞肺癌患者进行I/II期研究,确定患者的给药方案,未探索不同肿瘤类型;随后,对该人群进行了扩大样本的重点登记试验,由于研究开始时并没有明确结论EGFR突变为有益人群,因此未根据分子特征筛选人群,而是根据生物学特性进行筛选。收集所有患者的样本进行 EGFR 相关检测;上市后适用人群的进一步开发和给药方案的优化。可以看出,产品各个阶段的测试人群的选择是围绕其开发目标确定的。参考药物的选择也是如此。申办方明确表示,该产品的定位并不比现有的相同靶点药物差,因此未将化疗药物作为对照。当时可用的具有相同靶点的药物包括吉非替尼和厄洛替尼。虽然产品结构更接近厄洛替尼,但当时厄洛替尼在中国上市的时间并不长。中国患者使用的有效性和安全性数据较多,更有利于评价。申办方明确表示,该产品的定位并不比现有的相同靶点药物差,因此未将化疗药物作为对照。当时可用的具有相同靶点的药物包括吉非替尼和厄洛替尼。虽然产品结构更接近厄洛替尼,但当时厄洛替尼在中国上市的时间并不长。中国患者使用的有效性和安全性数据较多,更有利于评价。申办方明确表示,该产品的定位并不比现有的相同靶点药物差,因此未将化疗药物作为对照。当时可用的具有相同靶点的药物包括吉非替尼和厄洛替尼。虽然产品结构更接近厄洛替尼,但当时厄洛替尼在中国上市的时间并不长。中国患者使用的有效性和安全性数据较多,更有利于评价。当时厄洛替尼在中国上市的时间并不长。中国患者使用的有效性和安全性数据较多,更有利于评价。当时厄洛替尼在中国上市的时间并不长。中国患者使用的有效性和安全性数据较多,更有利于评价。
而且由于缺乏吉非替尼和厄洛替尼的头对头研究,目前还无法判断两者的优劣,所以最终选择吉非替尼作为对照的非劣效性研究。例如,如果正在开发靶向EGFR单一靶点的药物,那么当使用相同靶点药物的患者已被确认具有EGFR突变作为有益人群时,为该人群选择非突变人群似乎是不合适的。控制的选择 它不仅限于吉非替尼。更重要的是,目前针对非小细胞肺癌的EGFR单靶点药物有3个,多个多靶点的药物也已获批或将获批用于非小细胞肺癌领域。该领域新药开发市场似乎并不乐观,建议调整研发策略。4. 结论 小分子靶向药物是目前抗肿瘤药物研发的主流,市场上有多种产品。十年来,我们也积累了一些成功的经验,形成了一定的发展模式。例如,加强和完善早期探索性研究,在研究中尽可能多地收集信息;尽可能采集生物样本,尽早开展与疾病疗效相关的生物标志物的检测鉴定;基于生物标志物筛选,有可能获得优势人群;根据PK/PD分析确定可能产生疗效的剂量/程序(抑制靶点的有效浓度);根据前期研究结果及时调整研发计划,灵活多变的早期研究模型有利于获得更准确的目标人群定位和更准确的剂量,降低后续研究失败的风险。
借鉴市场上已有产品的成功经验具有事半功倍的效果。但是,任何产品的临床研发都无法复制,也没有固定的开发模式必须遵循。申办方仍需根据产品自身特点和实际情况制定合理可行的临床研究计划和策略,并注意使用现有的沟通交流程序,尽早征求管理层意见。此外,我们需要清醒地看到,近年来小分子靶向抗肿瘤药物的研发呈爆炸式增长。至少有数百种药物正在开发中。可以想象,未来5-10年,市场将非常拥挤。此类药物在项目的开发和审批中需要冷静理性。尽管生物标志物研究蓬勃发展,但与疗效和预后相关的明确确定的生物标志物毕竟有限。克唑替尼和埃克替尼同时开发生物标志物并不总是可能的。诊断试剂也存在困难,基于生物标志物的临床研究模型仍难以推广。细分具有某些分子特征的患病人群,导致病例难以入组,市场份额像孤儿药一样萎缩。它还需要制药公司和研究人员之间的合作,探索更多创新的临床研究模式。参考文献:[1]。Pham,.FDA 药物:您需要知道的。药物,2012,62-71. [2]。苏打 M、Choi YL、M 等。非细胞肺中的 EML4 -ALK 基因。2007 年 8 月 2 日;448(7153): 561-6. [3]. L.Kwak, et al. in Non-Cell Lung. New of .Oct 28,2010.363(18): 1693-1703 [4]. 陈晓元. 中国小分子靶向抗癌药物现状分析. 中国新药杂志, 2011, 20(17):1615-1619.@ >
免责声明: 本站关于疾病和药品的介绍仅供参考,实际治疗和用药方案请咨询专业医生和药师。






微信扫码◀
免费咨询电话
